Хвороба Гюнтера
Хвороба Гюнтера — одна з форм еритропоетичної порфирії, за якої збільшується утворення порфірину еритроїдними клітинами в кістковому мозку внаслідок недостатності уропорфіроген-III-косинтетази.
| Хвороба Гюнтера | |
|---|---|
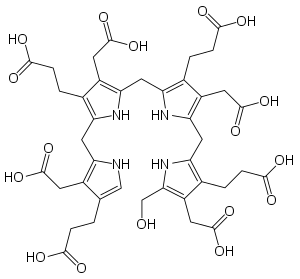 Гідроксиметилбілан, попередник уропорфіриногену III. Гідроксиметилбілан, попередник уропорфіриногену III. | |
| Спеціальність | ендокринологія і дерматологія |
| Класифікація та зовнішні ресурси | |
| МКХ-10 | E80.0 |
| OMIM | 263700 |
| DiseasesDB | 3048 |
| eMedicine | derm/145 |
| MeSH | D017092 |
Загальна характеристика
Перші прояви спостерігають на першому році життя, іноді через 4-5 років після народження.
Етіологія
Хвороба Гюнтера виникає внаслідок мутацій в гені, що кодує фермент уропрофіроген-3-синтетазу (UROS), та що розміщений в 10-й хромосомі (10q25.2-q26.3) людини[1].
Патогенез
Поряд з нормальними еритроцитами розвивається патологічний клон, в структурі якого накопичується багато уропорфиріну.
Клінічні ознаки
Хрящова тканина (носа, вух тощо, а також повік) запалюється та покривається виразками, що призводить до подальшої деформації. Відбувається зараження крові. Тому пацієнт потребує частого переливання крові, адже після контакту з сонячними променями відбувається згущення та зараження. Щоб лікувати хворобу Гюнтера, слід обрати донора кісткового мозку.
Примітки
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. с. 526. ISBN 0-7216-2921-0.